The National Medical Products Administration (NMPA) issued the Implementation Measures for Drug Regulatory Data Protection (for Trial Implementation, Draft for Comments) (“Implementation Measures”) and the Drug Regulatory Data Protection Working Procedures (Draft for Comments) (“Working Procedures”) on March 19, 2025. This review summarizes and comments on key provisions to assist stakeholders in formulating feedback to the NMPA.
Please click here to read the PDF version.
Highlights
The highlights of the Implementation Measures and the Working Procedures are as follows:
1. China’s regulatory data protection (“RDP”) is nearing its point of no return in terms of its implementation. In addition to patent litigation, pharmaceutical companies can rely on regulatory data protection to maintain their market share because:
- The chemical entities eligible for RDP have been significantly expanded,
- Biological products are included in the RDP, which benefits R&D on cutting-edge biological products.
We estimate that China’s RDP regime, if implemented, will significantly encourage innovative drug R&D.
2. Various measures have been taken to encourage international multicenter clinical trials to be conducted inside China even for drugs only marketed outside China. By having these drug trials inside China, it should encourage multinational pharmaceutical companies to simultaneously apply for marketing authorization in China for these innovative drugs.
3. Drug test data becomes a valuable, tradable and legally protected property right, which benefits brand-name drug companies and encourages the R&D of innovative drugs.
4. NMPA adopts the international mainstream “non-acceptance” and “non-approval” model. Hence, generic drug companies can reasonably arrange generic drug R&D plans based on the patent protection period and the RDP period.
Expanded RDP Regime
The Implementation Measures grant RDP based on drug registration classification, departing from China’s previously drafted RDP documents.
Chemical Drugs
RDP coverage extends beyond Class 4 Chemical Drugs to include all chemical drug categories, with separate RDP eligibility for different indications[1].
- Class 1 Chemical Drugs (Globally Innovative Drugs)
Core Protection: 6-year RDP period to prevent “free-riding” by generics.
The Implementation Measures indirectly define “new chemical ingredient” through the Chemical Drug Registration Classification and Application Material Requirements (No. 44 of 2020), i.e., “compounds with new clear structures and pharmacological effects”, which basically aligns with the definitions under other major jurisdictions, such as “new chemical entity”[2] under the U.S. Code of Federal Regulations. However, from the perspective of ensuring that laws are clear and accurate, we still recommend that the NMPA separately and clearly define “new chemical ingredients” under the Implementation Measures.
- Class 2 Chemical Drugs (Improved Drugs)
3-year RDP period: covers incremental innovations (e.g., new formulation, new indication) because such improvements satisfy the requirement of “origination of which involves a considerable effort” under Article 39.3 of the Agreement on Trade-Related Aspects of Intellectual Property Rights (“TRIPS Agreement”).
- Class 3 Chemical Drugs (Domestic Generics of Overseas-Marketed Drugs)
One of the most significant innovations of the Implementation Measures is that RDP can be granted in China for Class 3 Chemical Drugs.
Rationale: domestic generic drugs, which copy brand-name drugs that are marketed outside China, are required to conduct certain clinical trials when applying for marketing authorization in China according to the “Clinical Technical Requirements for Drugs Marketed Overseas but Not China-Marketed” (No. 29 of 2020).[3]
The Implementation Measures impact fully qualified Class 3 Chemical Drugs, i.e., generic drugs that copy brand-name drugs that are already marketed overseas, but neither the brand-name drugs nor their generic drugs having been marketed in China. Those drugs can seek RDP when they receive regulatory clearance, which will encourage domestic generic drug companies to develop fully qualified Class 3 Chemical Drugs.
For other Class 3 Chemical Drugs R&D, subsequent generics may obtain test data licenses to avoid redundant clinical trials.
For the marketing authorization holders (MAH) for overseas-marketed drugs, the Implementation Measures will indirectly force them to apply for the marketing authorization of the overseas-marketed brand-name drugs in China as soon as possible. For example, if the U.S. Food and Drug Administration (FDA) approves a U.S. MAH’s new drug, that MAH’s best business strategy in the China market is to apply for the marketing authorization of the new drug in China as soon as possible via the Class 5 Drug approval pathway and to apply for RDP. The sooner the MAH receives the China market authorization, the less room Chinese generic drug companies will have to market generic versions of the new drug. They will be left with no choice but to market their generic versions through the Class 4 Chemical Drug approval pathway.
- Class 5 Chemical Drugs (Overseas-Marketed Drugs Seeking China Marketing)
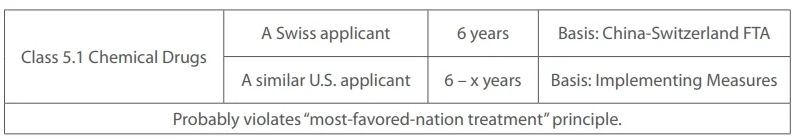
Innovative overseas-marketed drugs seeking China approval: 6 years minus x.
Improved overseas-marketed drugs seeking China approval: 3 years minus x.
“X” represents the time gap between the China marketing-application date and the overseas marketing date.
Example:
Take the Doptelet® developed by AkaRx, Inc. as an example:[4]
- Overseas marketing date: May 21, 2018 (approved by FDA)
- Domestic marketing-application date: December 21, 2022 (Category 5.1 Drug)
In this example, X is December 21, 2022 – May 21, 2018 = 3 years and 7 months.
And the corresponding RDP period is 6 years – 3 years and 7 months = 2 years and 5 months.[5]
By introducing variable “x”, the Implementation Measures indirectly encourages domestic and foreign pharmaceutical companies to conduct international multicenter trials during the drug R&D stage and to consider simultaneous marketing in China when applying for overseas marketing so that they can maximize the RDP period to create advantages for their drugs in China.
Nevertheless, this X variable may create issues with the “most-favored-nation treatment” principle under the TRIPS Agreement, Article 4, which mandates that any advantage, favor, privilege or immunity granted by a member to the nationals of any other country will immediately and unconditionally apply to the nationals of all other members. For example:
Biological Products
All major jurisdictions have included biological products in their own RDP regimes because of insufficient patent protection over biologics.[6]
The Implementation Measures expressly expand the scope of the RDP regime from new chemical entity drugs to biological products. This expanded scope encourages and promotes the R&D of high-end biological products such as CAR-T products, antibody products, mRNA vaccines, etc.
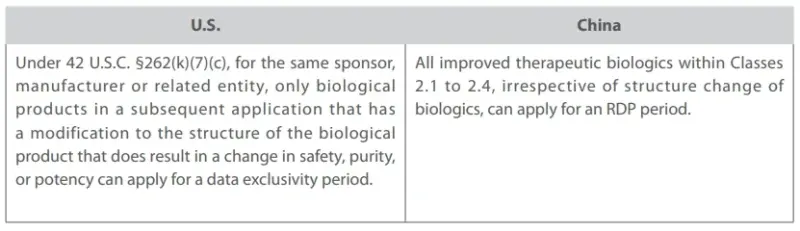
As for biological product RDP, another highlight of the Implementation Measures is the relaxed restrictions for Class 2 Improved Biological Products. Under the Implementation Measures, sponsors, manufacturers and other related entities that have obtained marketing authorization for innovative therapeutic biological products can apply for RDP in China for their improved biological products as long as their products are “improved”, as shown in the following comparison table.
Take Vyvgart developed by Argenx BV as an example[7].
Its first global marketing date is December 17, 2021 (approved by FDA), and its data exclusivity period is from 2021 to 2033. During this time, Argenx BV improved it to Vyvgart Hytrulo (FDA approved on June 20, 2023).
According to the Implementing Measures, Argenx BV (through Zai Lab as the licensee) can obtain a three-year RDP period for the Vyvgart Class 2.2 improved biological product “Vyvgart Hytrulo” (new indication, JXSS2400042) approved by the NMPA on November 5, 2024.
In considering potential impacts of this rule on the cross-border licensing transactions of biologics, we may see that when a licensee makes improvements, such as developing new indications for the licensed biologics, the licensee can obtain certain milestone payments from the licensor because it meets the requirements for obtaining RDP in China; and when the licensee makes structural changes to the biologics and produces safety, purity and efficacy improvements, the licensee can obtain a higher amount of milestone payments than the simple new indication improvement because it can apply for RDP and data exclusivity in the U.S. and China simultaneously.
Other Expectations for the Implementation Measures
Drug test data will become a valuable, tradable and legally protected property right
Unlike the “data exclusivity model” adopted by the U.S., the E.U., Switzerland and other developed countries, China has adopted the “anti-unfair competition model” proposed in the TRIPS Agreement, Article 39.3.
Under this “anti-unfair competition model”, the brand-name drug company, including the marketing authorization holder of the first Class 3 Generic Chemical Entity Drug, can license its protected test data to the generic drug company at the end of the RDP period or at other appropriate times. This rule establishes that the test data of brand-name drugs is a valuable, tradable and legally protected property right, which will undoubtedly increase the value of brand-name drugs and further encourage the R&D of innovative drugs.
Pharmaceutical companies will be encouraged to consider simultaneous marketing applications for innovative drugs in China
Under the Implementation Measures, the NMPA aims to increase new drugs in China by encouraging multinational pharmaceutical companies to conduct international multicenter clinical trials in China during the drug R&D stage and to apply for marketing applications in China simultaneously or as early as possible. China’s clinical trial industry stakeholders, such as CROs, will also stand to benefit because necessary clinical trials are needed.
NMPA adopts the phased protection model of “non-acceptance” and “non-approval”
In terms of the international RDP regime, the mainstream practices are “non-acceptance” and “non-approval”. Under the Implementation Measures, the drug administration authority will refuse to accept the marketing application for a generic drug before the final year of the RDP period while a marketing application can be submitted within one year before the RDP period expires even though the drug administration authority will not grant the marketing clearance until the RDP expires.
NMPA should consider extending the data protection period for innovative therapeutic biological products
Under the current version of the Implementation Measures, the 6-year RDP period may be too short for innovative chemical entity drugs and innovative therapeutic biological products. In particular, for biological products, biological products, empirical studies from the U.S. show that the remaining patent life for biological products is only 10.3 years (contingent PTE inclusive).[8] Considering that the R&D cycle of innovative therapeutic biological products is much longer, the cost is much higher, and the risk of failure is much greater, the U.S. and the E.U. have all set an RDP period of more than 6 years for biological products. We recommend that the NMPA likewise consider extending the data protection period for innovative therapeutic biological products.
Conclusion
New drug development is an adventure with large investments, long cycles and high risks. Long-term and continuous clinical trials are required to acquire data on drug efficacy, safety and quality. Hence, RDP protection, in addition to patent protection, provides another regulatory buffer to encourage new drug R&D and protect the resulting products even after their long development times.
Empirical studies from the U.S. and the E.U. have shown that RDP can effectively promote the development of innovative drugs and benefit public health, RDP can block and delay the entry of generic drugs to a certain extent, creating concerns about high drug prices making those drugs unaffordable for ordinary people. And despite the belief that generics and biosimilars reduce prices through competition, according to an empirical study from Finland in 2024, biosimilars fail to reduce prices as imagined. Instead, prices depend more on a country’s pricing controls and reimbursement system.[9]
Based on these realities, for China to achieve the dual vision of balancing the encouragement of new drug R&D and improving public health through accessibility to and affordability of new drugs, China’s medical, pharmaceutical, and insurance authorities will need to consider the interchangeability of brand-name and generic drugs and to continuously reform its medical insurance system.
- Article 5, Paragraph 4 of the Implementation Measures. ↑
- 21 CFR § 314.108(a). ↑
- Clinical Technical Requirements for Drugs Marketed Overseas but Not China-Marketed: (Generic Drugs Marketed Overseas and Domestically) The clinical trial requirements for generic drugs that are marketed overseas but not marketed domestically shall be determined after comprehensive consideration of the clinical evaluation results of the original drug and the formulation of the drug: … Based on the clinical evaluation results of the original drug, the requirements for conducting necessary clinical trials in the Chinese patient population are consistent with those for the original drug. Since it is difficult to obtain complete clinical test data for the original drug, which may affect the full clinical evaluation of the original drug, it is usually necessary to conduct necessary clinical trials to support the safety and effectiveness evaluation of generic drugs for Chinese patients… [emphasis added]. ↑
- As the Implementation Measures are still in the comment period, they are not legally binding. Therefore, this example is only for illustrative purposes and is not intended as a legal opinion. ↑
- The Implementation Measures do not specify the benchmark calculation date for the overseas marketing date. The local approval time of FDA is Maryland time (GMT-4). The local approval time of EMA is Amsterdam time (GMT+1). The local approval time of NMPA is Beijing time (GMT+8). ↑
- For example, the E.U. granted biological products an “8+2+1” year data exclusivity in Directive 2001/83/EC and Regulation (EC) No 726/2004, and the U.S. granted biological products a 12-year data exclusivity under the Biologics Price Competition and Innovation Act (BPCI Act) of 2010. ↑
- As the Implementation Measures are still in the stage of soliciting opinions, they are not legally binding. This example is only for illustrative interpretation and is not intended as a legal opinion. ↑
- C. Benson Kuo and Frances Richmond, Use of patent term extensions to restore regulatory time for medical devices in the United States, 21 Expert Rev. Med. Devices 527, 528-9 (2024) and Oliver J. Wouters et al., Differential Legal Protections for Biologics vs Small-Molecule Drugs in the US , 332 J. Am. Med. Ass’n 2101, 2103 (2024). ↑
- Sanna V. Luukkanen et al., The Price and Market Share Evolution of the Original Biologics and Their Biosimilars in Finland, 36 BioDrugs 537, 546 (2022). ↑