引言
为响应国务院在2024年《关于全面深化药品医疗器械监督改革促进医药产业高质量发展的意见》中提出的药品试验数据保护期的改革方向,国家药品监督管理局于2025年3月19日发布了《药品试验数据保护实施办法(试行,征求意见稿)》((“《实施办法》”)和《药品试验数据保护工作程序(征求意见稿)》(“《工作程序》”),这标志着中国政府再一次尝试将药品试验数据保护制度真正落地,以及中国政府促进医药行业高质量发展的决心。就行业所关心的问题,我们对《实施办法》和《工作程序》进行了如下解读和评论,供业内参考并向国家药品监督管理局反馈相关意见。
新规亮点总结
本次《实施办法》和《工作程序》有如下亮点:
1. 药品试验数据制度落地箭在弦上。在漫长的专利诉讼保护之外,药企可以依赖数据保护制度保持自身市场占有地位,进而整体鼓励创新药研发;
- 化学药品试验数据保护范围显著扩大;
- 生物制品纳入药品试验保护范围,利好高端领先生物制品研发;
2. 多种举措鼓励境外已上市境内未上市药品在药物研发阶段在华开展国际多中心临床试验,并鼓励创新药物同步在华申请上市;
3. 药品试验数据成为一种有价值,可交易并受法律保护的产权,利好原研药研发企业,鼓励创新药研发;
4. 采国际主流的“不受理”和“不批准”的分阶段数据保护模式,仿制药企业可以根据专利保护期和数据保护期合理安排仿制药研发计划。
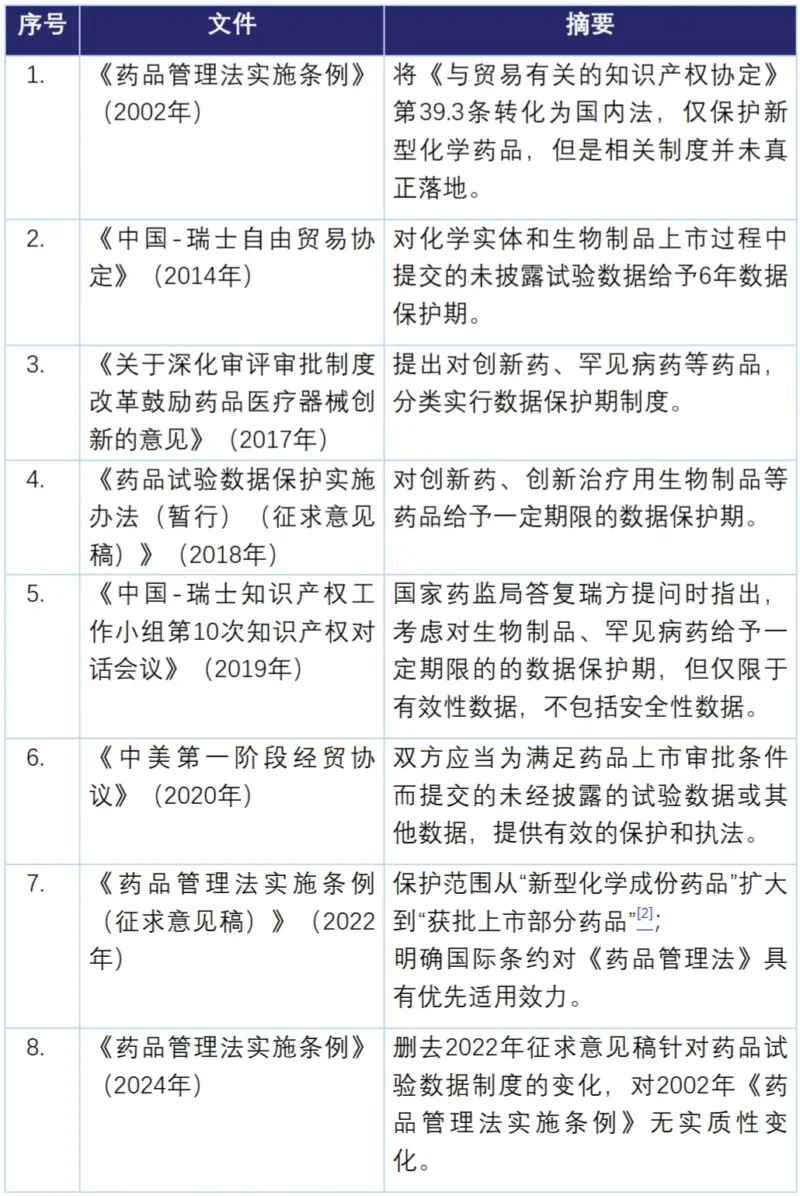
中国药品试验数据制度的历史发展
药品试验数据保护制度最早起源于美国1983年的《孤儿药法案》(Orphan Drug Act),并在1984年的《哈奇-韦克斯曼法案》(Hatch-Waxman Act)推广到一般新型化学药品(NCE Drugs)。在1986年开始的乌拉圭回合谈判中,以美国、欧盟、瑞士为代表的发达国家在谈判中力推将药品试验数据作为一项知识产权保护,并最终写进了《与贸易有关的知识产权协定》(TRIPS Agreement)。
《与贸易有关的知识产权协定》明确药品试验数据是一种知识产权[1],且第39.3条要求各WTO成员国对“利用新型化学实体”的药品在上市后给予一定期限的数据保护期。
自中国2001年加入世界贸易组织(WTO)以来,中国的药品试验数据制度长期处于有规定无细则的“悬空状态”:
药品试验数据保护范围扩大
相较于2018年的《药品试验数据保护实施办法(暂行)(征求意见稿)》,本次《实施办法》直接按照药品注册分类标准授予试验数据保护期,我们将分别按照不同药品注册分类标准进行评述。
1. 化学药品
本次《实施办法》将药品试验数据保护范围扩大到了除第4类(境内已上市药品的仿制药)以外的所有化学药品,且不同的适应症均可申请单独的数据保护[3]:
(1) 第1类化学药(境内外均未上市的创新药)
创新药的试验数据保护是试验数据保护制度的题中之意,试验数据保护可以作为一定形式的“垄断”来防止仿制药“搭便车”,保证原研药研发者在药品上市后一定期限内的可观收益,从而鼓励创新药的研发。
本次《实施办法》并没有明确何为“新型化学成份”。仔细观察,《实施办法》是通过《化学药品注册分类及申报资料要求》(2020年44号)间接定义了1类化学药的“新型化学成份”,即“含有新的结构明确的、具有药理作用的化合物”。该定义与其他司法辖区对此的定义,例如美国联邦法规第21章(21 Code of Federal Regulations)下的“新型化学实体(new chemical entity)”,保持了基本一致[4]。不过,从法条须保持明确和准确的角度,我们仍然建议国家药品监督管理局在《实施办法》下单独且明确地定义“新型化学成份”。
(2) 第2类化学药(境内外均未上市的改良型新药)
相较于1类创新药,2类改良药的创新程度不足,但是其研发过程中产生的试验数据也满足了《与贸易有关的知识产权协定》第39.3条下规定的“付出可观努力取得”的要求。本次《实施办法》参考了主流司法辖区的通行做法,给予2类改良药3年的数据保护期。
(3)第3类化学药(境内申请人仿制境外上市但境内未上市原研药品的药品)
本次《实施办法》最大的创新点是将3类仿制药的试验数据也纳入试验数据保护范围。
3类仿制药的试验数据的保护原因在于,依据《境外已上市境内未上市药品临床技术要求》(2020年29号)的规定,境外已上市境内未上市的仿制药品在中国申请上市时需要开展一定的临床试验[5]。
就本次《实施办法》对3类仿制药的影响而言,真3类仿制药(境外已上市,境内原研和仿制均未上市)获批上市时可以寻求试验数据保护,这将鼓励境内仿制药企业对真3类仿制药研发。对后续的二仿、三仿等3类仿制药而言,研发企业可以以一定对价向首仿3类药企寻求首仿制药的数据依赖许可,以节省开展必要临床试验的高额成本,从而鼓励后续仿制药的研发,提高药品丰富度并产生药品竞争效果。
而对于已境外上市的药品上市许可持有人而言,本次《实施办法》将3类仿制药纳入试验数据保护还将会倒逼其早日通过各种方式(包括但不限于通过其中国子公司、通过中国被许可方)尽早申请境外已上市原研药在中国上市。例如FDA已批准了一款新化学药品A,上市许可持有人是美国甲公司,那么对于甲公司在中国市场的商业策略而言,较为理想的商业布局是将A药品尽快在中国依照第5类化学药路径申请上市并申请数据保护期。如此以来,该等药品A就属于境内已上市的药品,中国仿制药公司就只能按照第4类标准申请上市。
(4) 第5类化学药(境外上市的药品申请在境内上市)
本次《实施办法》另外一个显著的创新点是给予第5.1类药 6-x年(境外上市的原研药品申请在境内上市)或3-x年(境外上市的改良型药品申请在境内上市)的试验数据保护期,其中x代表境内受理时间与境外上市时间的时间差。
以AkaRx, Inc研发的阿伐曲泊帕片(商品名称:Doptelet®)为例[6]
其境外上市时间为:2018年5月21日(由FDA批准上市)
境内申请上市时间为:2022年12月21日(第5.1类化学药)
则x为2022年12月21日-2018年5月21日=3年7个月
则相应的药品试验数据保护期为2年5个月[7]
变量x在第5.1类药申请上市过程中起到了关键作用:境外上市获批时间与境内申请上市时间间隔越短(x越小),其在中国的数据保护期越长,从而变相鼓励了境内外药企在药物研发阶段开展国际多中心试验,并考虑药物境外申请上市时的中国同步上市,这样方可将其药品在中国的数据保护期以及市场利益最大化。
不过,本次《实施办法》对第5.1类药品的变量x创新可能会产生《与贸易有关的知识产权协定》下的“最惠国待遇”问题。《与贸易有关的知识产权协定》第4条确立了最惠国待遇原则(任何成员国给任何成员国的任何优惠、特权,都应当立即且无条件地不低于前述标准,给予其他任何成员国)。本次《实施办法》就最惠国原则而言,我们可以预见一个瑞士申请人依照《中国-瑞士自由贸易协定》可以就其5.1类药品获得6年数据保护期,但是一个美国申请人依照《实施办法》就其5.1类药品只能获得6-x年数据保护期,这可能违反WTO规则下的“最惠国原则”规定。对此潜在问题,我们期待国家药品监督管理局进一步考虑,并在最终稿对此进行澄清。
2. 生物制品
相比于小分子化学药,生物制品具有分子量大,结构复杂,生物学技术制成等特点,这些特点导致了专利法对生物制品保护的不足(例如可能无法满足“创造性/creativity”要求而无法获得专利保护,等同侵权原则也无法完全保护生物制品方法专利[8])。有鉴于此,各主要司法辖区均将生物制品的试验数据纳入药品试验数据保护制度[9]。
本次《实施办法》将试验数据保护范围明确扩大到生物制品,不再局限于新型化学药品。这一举措无疑是顺应国内医药产业研发现状,进一步鼓励和推动高端生物制品(例如CAR-T产品、单抗产品、mRNA疫苗等)的研发。同时,这也是对《中国-瑞士自由贸易协定》要求的生物制品试验数据保护的落地,是国家落实《对外关系法》下“善意落实国际条约义务”的体现[10]。
在生物制品试验数据保护下,本次《实施办法》另一亮点在于第2类改良型生物制品,极大放宽了对改良生物制品数据保护范围的限制:从目前的《实施办法》来看,即便是已经取得创新治疗用生物制品上市许可的申办方、制造商等相关实体,只要取得了符合要求的改良,均可以在中国申请试验数据保护(如下表对比所示)

以Argenx BV研发的“卫伟迦/Vyvgart”为例[12]:
其在全球首次上市时间为2021年12月17日(FDA批准),其数据独占期为2021年至2033年。在此时间内Argenx BV对其做出改良Vyvgart Hytrulo(FDA于2023年6月20日批准)。
根据《实施办法》,Argenx BV(通过再鼎药业作为被许可人)在国家药品监督管理局2024年11月5日批准的Vyvgart第2.2类改良药“卫力迦/Vyvgart Hytrulo”(新增适应症,JXSS2400042)可以依照《实施办法》,获得3年的试验数据保护期。
我们理解此项做法目的在于鼓励已取得创新治疗用生物制品中国上市许可的申办方等,通过其本身或者通过其被许可方,持续对该已上市产品进行改良(无论该等改良是新增适应症,还是结构变化导致的安全、纯度和效力变化)。这一举措无疑是增大了原有创新治疗用生物制品的商业价值,并进一步鼓励国内外药企之间开展针对1类创新生物制品的许可和并购交易。特别的,对于生物制品跨境许可交易而言,未来我们可能看到:当被许可方(Licensee)针对许可的生物制品做出了例如新增适应症、给药途径等非结构变化导致的安全、纯度和效力改良,因其满足获得中国数据保护期的要求,被许可方可以从许可方(Licensor)处获得一定的里程碑付款;而当被许可方做出了生物制品结构变化并产生安全、纯度和效力改良,因其可以同时在美国和中国申请数据保护期,那么被许可方可以获得相比单纯新增适应症改良而言金额更高的里程碑付款。
《实施办法》的其他亮点与进一步期待
1. 药品试验数据成为一种有价值、可交易并受法律保护的产权
与美国、欧盟、瑞士等国家的“数据独占期模式”不同,中国在药品试验数据保护制度上采用了乌拉圭回合谈判中发展中国家所提出的,也是最后被《与贸易有关的知识产权协定》采纳的“反不正当竞争模式”。
“反不正当竞争模式”最大的特点在于其并不排斥数据保护期内仿制药的上市,只要原研药企业同意仿制药企业依赖其试验数据即可。因此,原研药公司(包括首个第3类仿制化学药企业)完全可以在数据保护期的末尾或者其他合适时机,将其受保护的试验数据以一定的对价许可给仿制药公司使用(特别是考虑到仿制药公司获得一定的试验数据也并不能保证仿制药的成功研发)。当原研药的药品试验数据成为一种有价值、可交易并受法律保护的产权,这无疑会增加原研药及其研发企业的价值,鼓励创新药的研发。
2. 鼓励药企考虑创新药品在中国的同步上市申请
长时间以来,由于国内强大的仿制药产业,部分境外药企担心其药品价值流失,常常选择推迟将最新研发的产品在中国上市。本次《实施办法》下,我们可以看到国家药监局通过制度建设,在制度上鼓励境外药企在药物研发阶段在中国开展国际多中心临床试验,以及同步或者早日在中国申请新研产品上市申请,以进一步促进中国市场药品可及性。考虑到境内药品上市过程中部分必要临床试验必须在中国开展,涌来的境外药物/药品境内上市需求对中国的临床试验行业参与方,例如CRO等,无疑是利好消息。
3. “不受理”和“不批准”的分阶段保护模式
试验数据保护制度下的“不受理”和“不批准”是国际主流通行做法。《实施办法》第11条下,获得药品试验数据保护剩余1年前的时间段内,药品监督管理部门不受理仿制药的上市申请;数据保护期届满前1年内可以提交申请,但是药品监督管理部门不批准上市。其中,根据《工作程序》,不批准上市的中止评审理由为“药品试验数据保护”。仿制药企业可以根据这一条的规定,合理规划仿制药研发布局;对原研药公司而言,也可以根据这一条的规定确定合理的对仿制药企业数据依赖许可的时间。
4. 差异化数据保护期
目前《实施办法》下,创新化学药和创新治疗用生物制品的数据保护期限均为6年。特别的,就高端的创新治疗用生物制品而言,来自美国的实证研究表明[13],生物制品从专利申请到药品获批上市花费12.6年(中位数),剩余专利期7.4年(中位数),加上或有的平均专利延长期(PTE)年2.9年[14],生物制品的专利保护期仅为10.3年。考虑到创新治疗用生物制品的研发周期更长,成本更高,失败风险更大,并且生物制品的相关专利保护略显不足,美国和欧盟均设置了超过6年的生物制品试验数据保护期。因此,针对创新治疗用生物制品,有必要适当延长数据保护期限。我们建议国家药监局借鉴美国和欧盟政府部门所采用的盈亏模型并在结合中国当前药品研发实际情况的基础上进一步测算中国生物制品的合理保护期限。
结语
新药研发是一场风险高、投资大、周期长的冒险之旅。药物有效性、安全性和质量数据的获得需要长期推进和不断拓展的临床试验。因此药品试验数据保护,在专利保护之外,为新药研发提供另外一道监管屏障,以鼓励新药发展(例如,在管线许可交易中,通常情况下决定特许权使用费的支付期限的其中一个维度就是数据/市场排他期,中国药品试验数据保护制度不断落地和推进,从侧面也体现了国家政府部门对于鼓励新药研发和创新的方向和决心)。来自美国和欧洲的实证研究均表明,药品试验数据保护可以有效促进创新药研发并有益于公共卫生。当然,药品试验数据保护一定程度上阻断、推迟了仿制药进入,带来了对药品价格高、普通百姓无法负担的担忧。根据2024年一项来自芬兰的实证研究,仿制药(生物类似药)带来的价格竞争并非如想象般降低药品供应价格,药品价格的可及性更多取决于一国的药品价格管制和医保制度[15]。我们期待国家有关部门在推动药品试验数据保护制度落地的同时,考虑原研-仿制药互换性(interchangeability)以及医保制度的不断改革,以期实现平衡鼓励新药研发和提高公共卫生的双重愿景。
- Agreement on Trade-Related Aspects of Intellectual Property Rights, art.1.2. ↑
- 2022年的《药品管理法实施条例(征求意见稿)》第40条用语是“获批上市部分药品”,而之前的《药品管理法实施条例》相关用语是“新型化学成份药品” ↑
- 《实施办法》第5条第4款 ↑
- 21 C.F.R. § 314. 108(a). ↑
- 《境外已上市境内未上市药品临床技术要求》:(境内外仿制药品)对于境外已上市境内未上市药品的仿制药的临床试验要求,需结合原研药品临床评价结果及制剂学两个方面的因素综合考虑后确定:…基于原研药品临床评价结果,开展必要的中国患者人群临床试验的要求与原研药品一致。由于难以获得原研药品完整临床试验数据,可能影响对原研药品进行充分临床评价,故通常需开展必要的临床试验以支持仿制药用于中国患者的安全性和有效性评价… ↑
- 鉴于《实施办法》仍在征求意见阶段,不具有法律约束力。本例仅做示范性解释,不构成任何法律意见和观点。 ↑
- 注:本次《实施办法》未注明境外上市日期的基准测算日期,FDA当地批准时间为马里兰州时间(GMT-4),EMA当地批准时间为阿姆斯特丹时间(GMT+1),而NMPA当地批准时间为北京时间(GMT+8)。 ↑
- 例如Amgen Inc. v. Sandoz Inc. 923 F.3d 1023 (Fed. Cir. 2019), 华瑞同康生物技术(深圳)有限公司、南京诺尔曼生物技术股份有限公司侵害发明专利权纠纷 (2020)最高法知民终342号。 ↑
- 例如欧盟在Directive 2001/83/EC和Regulation (EC) No 726/2004年给予生物制品“8+2+1”年的数据独占期,美国在2010年的《生物制品价格竞争与创新法案》(BPCI Act)给予生物制品12年的数据独占期。 ↑
- 《对外关系法》第30条:国家依照宪法和法律缔结或者参加条约和协定,善意履行有关条约和协定规定的义务。国家缔结或者参加的条约和协定不得同宪法相抵触。 ↑
- 42 U.S.C. §262 (k)(7)(C) ↑
- 鉴于《实施办法》仍在征求意见阶段,不具有法律约束力。本例仅做示范性解释,不构成任何法律意见和观点。 ↑
- Oliver J. Wouters et al., Differential Legal Protections for Biologics vs Small-Molecule Drugs in the US, 332 J. Am. Med. Ass’n 2101, 2103 (2024). ↑
- C. Benson Kuo and Frances Richmond, Use of patent term extensions to restore regulatory time for medical devices in the United States, 21 Expert Rev. Med. Devices 527, 528-9 (2024). ↑
- Sanna V. Luukkanen et al., The Price and Market Share Evolution of the Original Biologics and Their Biosimilars in Finland, 36 BioDrugs 537, 546 (2022) ↑